中国战略新兴产业融媒体记者 艾丽格玛
“人类泛基因组参考图谱是我们已经等待了十年的一个里程碑,这是在测序技术和生物信息学方面取得了诸多创新后才得以达成的。”这是人类泛基因组参考联盟的主要研究者之一、德国海因里希海涅大学(Heinrich Heine University)的Tobias Marschall教授对近日重大成果的评价。
顶尖学术期刊《自然》近日同时发表了3篇人类泛基因组相关论文,另有1篇论文发表于《自然-生物技术》。在这一专题中,人类泛基因组参考联盟(Human Pangenome Reference Consortium,HPRC)提出了首个人类泛基因组参考草图的构建和使用方法,并报告了使用该图谱获得的两项新发现。
最新图谱结合了来自不同祖源的47个个体的遗传信息,比现有的单一参考基因组序列(GRCh38)新增1.19亿个碱基字母。该图谱为人类基因组提供了更完整的图像,也更能代表人类这个物种的遗传多样性,应用这一参考图谱能极大提高对人类基因组中变异体的检测。
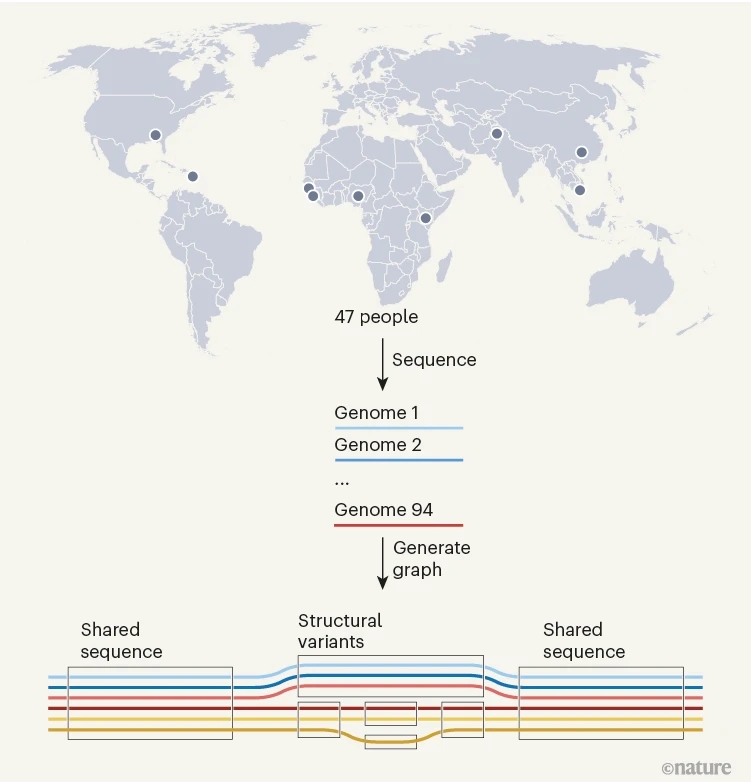
图:人类泛基因组参考草图。来自47位不同血统的人的基因组被用来生成这个草图。每个个体都生成了两个全基因组序列,这94个序列被比对以形成一个全基因组图,这在概念上类似于一张地铁地图。框中的区域表示在给定位点存在于一个或多个基因组中的序列,分支路径表示序列变异。这些图表揭示了大型基因组改变,称为结构变异,并可以轻松分析它们在不同个体之间的变化。来源:《自然》
早在去年,HPRC就在《自然》上阐释了泛基因组计划的目标和战略。而如今,HPRC的研究人员介绍,该项目还将继续采样,目标是纳入350名个体的基因组信息,尤其是目前还未纳入的人群代表,以便扩大多样性和增强不同人群之间的平衡性。他们计划在2024年发布人类泛基因组参考的最终版本,力求代表“人类”这个物种尽可能多的DNA序列。
泛基因组是什么?
每个人都是独一无二的,每个人的基因组也略有不同。人类基因组含有约31亿个DNA碱基对,平均来说,每个人与别人的基因组有大约0.4%的差异。而一个物种参考基因组的构建,往往是选取一个样本,以线性排列的方式进行呈现,但单个个体遗传信息是具有局限性的,研究者一直都在寻求能够完全包含一个物种所有的遗传变异信息,并使后续的基因组分析能够更有效率地参考基因组形式。
读懂人与人之间的这些特异的微小差异,可以更好地掌握一个人的健康与疾病状况,有助于疾病的诊断、治疗方法的选择、治疗结果的预测。
泛基因组(Pan-genome)的概念最早由Herve Tettelin等人提出,表示一个物种全部基因组成,包括核心基因(Core gene)、非必须基因(Dispensable gene)和个体特异性基因 (Strain-specifific gene)。其中,该物种所有样本共有的基因称为核心基因,一般与物种生物学功能和主要表型特征相关;仅在物种部分样本中存在的基因称为非必须基因,一般与物种对特定环境的适应性或特有的生物学特征相关,反映了部分物种的特性;某一样本特有的基因称为个体特异性基因,反映该个体的特异性状。
不过,由于时代的关系,泛基因组在不同类型的物种里面,定义上有细微的区别。2009 年,Li等人首次采用新全基因组组装方法对多个人类个体基因组进行拼接,发现了个体独有的DNA序列和功能基因,并首次提出了“人类泛基因组”的概念,即人类群体基因序列的总和。
此前已有的参考基因组序列,一个重要局限在于,它是由大约20名志愿者的遗传数据拼凑组成的一套基因组——实际上,大部分参考序列(约70%)实际上只来自其中一名志愿者。
“无论你对一个基因组的表征有多准确,都不能代表所有人类群体。”从一开始就参与人类基因组计划,也是人类泛基因组参考联盟一员的David Haussler教授解释说,“现在则是一个转折:不再是‘一个标准人类基因组’的基因组学,而是‘可用于所有人’的基因组学。”
在实际应用过程中,当科研人员或临床医生想要根据一个人的基因组找出与疾病相关的变异,通常情况下要将新测序的人类基因组数据和参考基因组进行比对,才能够获得个体、人群之间所存在的序列差异,然后再进行后续研究,比如寻找疾病的遗传起源、肿瘤的易感因子、药物开发的靶点、特定目的基因芯片的设计等,参考序列就是这个过程中最重要的标准参照物——它是一个基因组学的“参考坐标系”。由于基因组学的研究都需要围绕 “参考基因组” 来开展,因此它的作用和意义非凡。然而,人类群体中存在大量遗传变异,有些变异在某些特定人群(例如特定祖源)存在,在另一些人群中不存在。
因此,在临床使用基因组学信息——例如预测一种遗传病时,如果想要减少偏差,作为标准的参照物就不能只是某个单一人群的代表,而需要扩大其代表性。
此次发布的人类泛基因组参考纳入了完整的基因组序列,还拓展了多样性上的新维度,将来自47名志愿者的全基因组序列集合排列,能方便地分析基因组在个体之间的变化,反映人类物种内部的遗传多样性。
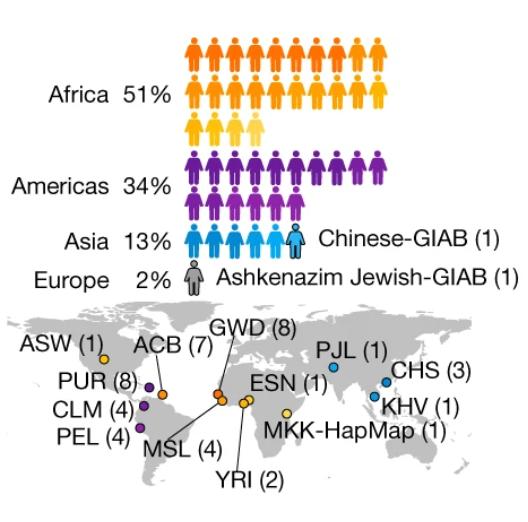
图:47名志愿者所属的样本亚群。来源:《自然》
新的泛基因组参考便提供了更多样化,也更准确的标准。研究人员通过复杂的算法,将组装完成的单个基因组序列集合编译为图形结构。
如果说过去的参考基因组是“一根线”,那么现在的人类泛基因组参考是多个基因组序列的“多线并行图”。在碱基完全相同的序列,它仍然是单线;而存在人群差异的序列部分,线条则从一根“分化”出铁轨般交错的多根线条。
突破来源于重要的前置技术的进步
2003年,人类基因组计划成功绘制了人类基因组90%以上的序列。但是,受到当时的测序技术所限,这版草图中留有许多空白,参考序列在此后的多年里不断更新、修复错误、填补空白。但直到2019年,人类基因组测序结果中仍有数百万个碱基位置上是空白的。
自2005年新一代测序出现以来,早期平台的读长较短,需要依靠强大的算法将重叠序列的短读数连接起来。然而,许多基因组庞大且复杂,这就给完整序列的生成带来了严重的障碍,也是导致基因组中出现缺口的原因。
而就在去年,基因组技术取得重大突破。
《Nature Methods》杂志将“2022年度科学方法”授予了“长读长测序技术”,这种测序技术能够检测长度在1000-20000个碱基或更长的DNA (或RNA)片段。这些片段通常来自“原生”分子,这些分子是直接从生物样本中提取出来进行分析的。而此前使用的大多数短读长测序技术只能检测50-300个碱基长度的片段。
使用长读长测序数据,组装基因组变得容易多了。打个比方来说,长度长测序就像是在用包含整个段落的文字来拼凑一本500页的小说一样——这些长段落提供了关于情节中重要事件的上下文信息,使得人们可以更方便地将它们按照正确排序来重建;而短读方法就像是用一些支离破碎的词组来拼凑一本具有完整情节的书籍。使用长读解决方案创建基因组,需克服的障碍比使用短读解决方案更为简单,需要的计算步骤更少、更简单。
长读长和超长读长测序技术的迅猛发展,使得研究者读取生物样本的DNA时可以一次性解码几千甚至百万个碱基对,然后通过专门的算法将这些DNA长片段组装成更完整的基因组序列。
就在这一基础上,由美国国家人类基因组研究所、加州大学圣克鲁兹分校、华盛顿大学等组成的国际科学团队“端粒到端粒”联盟(T2T,Telomere-to-Telomere,T2T也加入了HPRC)建出了第一个完整的人类参考基因组,相关研究结果此前已在《科学》杂志上发表的论文中公布。
为什么泛基因组图谱如此重要?
过去,在使用单一参考基因组的时候,人类基因组中存在的结构变异有70%以上难以识别。而现在,基于更全面的泛基因组参考图谱进行基因组分析,结构变异的检测率可以提高104%,研究人员有机会在未来将结构变异与疾病更好地联系起来。除了结构变异的检测率提高外,检测较小的遗传变异(例如只有一个或数个碱基差异)时,使用泛基因组参考的准确性也有34%左右的提高。
最新的泛基因组参考图谱还有一项重大突破。我们的染色体是成对存在的,一套遗传自母亲,一套遗传自父亲,而泛基因组参考包括的单倍型信息——来自47个人的94套基因组序列,可以在分析一个人的基因组时更准确地区分出来自父母的不同染色体。这也将帮助我们更好地理解各种基因和疾病的遗传方式。
总的来说,使用更加完整的泛基因组参考图谱有三个非常明显的优势。
首先,当对患者样本进行测序和分析时,一个包含人类遗传多样性的更完整的参考基因组将产生更少的模糊映射和更准确的全基因组范围内的拷贝数变异分析,这将改善基因诊断和变异的功能注释。
其次,这一资源将有助于发现疾病风险等位基因和以前未观察到的罕见变异,特别是在标准短读长测序技术无法获得的区域,例如那些重复扩增位点,通过长读长测序对这些位点进行解析,提高了基因分型的能力,使得通过全基因组关联研究和定量性状位点鉴别方法去发现新的遗传关联成为可能。
第三,完整的全基因组代表了人类基因变异如何被发现并被鉴定的根本性转变,从简单地将患者序列比对到一个参考基因组上,发展为通过构建分阶段的基因组组合,并将它们与参考图表比对,以在碱基对水平上精确定位所有的基因差异。
圣路易斯华盛顿大学的王艇教授是该项目的主要研究者之一,他介绍:“新的泛基因组参考能更准确地发现和评估人类遗传变异,特别是结构变异。人类遗传学和基因组医学的几乎所有领域都可以因此受益。例如,鉴定与人类疾病相关的遗传变异将更加敏感和具体,从而直接改善疾病诊断和治疗。新的参考基因组还为研究遗传变异的功能后果奠定了基础。”
例如,在2022年9月15日,上海交通大学朱正纲、陈红专、韦朝春及于颖彦共同通讯在《Nature Communications》发表题为“Pangenomic analysis of Chinese gastric cancer”的研究论文,该研究报告了使用人类泛基因组分析的自动化流程为185对深度测序数据(370个样本)构建胃癌泛基因组,并在全基因组水平上表征基因存在-缺失变异(PAVs)。基因ACOT1、GSTM1、SIGLEC14和UGT2B17被鉴定为胃癌群体中高度缺失的基因。预测了一组来自未比对序列的GRCh38基因。该研究还鉴定出染色体9q34.2上的预测基因GC0643为肿瘤抑制基因。研究成果为解析中国人胃癌高发的分子遗传学背景提供了重要参考。
此外,对于细菌的泛基因组来说,相关研究有助于我们研究菌株内的遗传多样性,深入了解菌株致病性、毒力和耐药性差异的根本原因,而且还可以帮助我们预测新菌株的危害性。这对于人类健康来说有实际的意义;对于真核生物的泛基因组特别是植物泛基因组研究来说,意义也很大。据了解,现在已经有多个农作物的物种泛基因组被构建出来,包括水稻、西红柿、大豆、白菜、西兰花和向日葵等。构建这些农作物泛基因组,通常都是为了了解作物基因组上的哪些突变会影响农作物的表型,从而改善育种。通过泛基因组分析,可以用来揭示农作物表型和特定基因型的关联,反过来就可以帮助我们定向地进行作物育种和基因改良,然后创造出更高产、更具抗病性、更长保质期和口感更好的作物。
目前还有哪些问题尚未解决?
研究称,GRCh38是目前已知的人类基因组序列的一个标准版本,它由一小部分人类基因组测序数据组成,其中包含许多已知的人类基因组变异和SNP(单核苷酸多态性)信息。GRCh38是人类基因组研究的重要工具,可以用来识别、定位和注释基因,以及进行遗传变异分析和比较基因组学研究。
但是,由于人类基因组是高度复杂的,存在很大的个体差异和变异形态,因此仍需要更深入的研究和探索来完善我们对其的理解。
在当前的GRCh38版本中,有210 Mb(兆碱基)的DNA序列段没有被完整地测序,或无法确定其序列,其中151 Mb的区域完全未知,而59 Mb的区域是通过计算机模拟得到的预测序列。
这种情况会造成相关研究的数据偏差,也意味着在人类基因图谱中,依然有很多区域是我们尚未可知的。因此,我们仍然需要不断完善它。
可以说,这些成果还只是人类泛基因组研究发展中的一个过渡阶段。研究人员Arya Massarat强调了目前成果的重要性,但也表示,还需要持续改进以克服现有不足,比如,更多样化的取样。“这将帮助我们理解促成生理和临床特征的遗传变异,并为全球健康事业做出贡献。”(综合编辑)